【综述】晶状体后圆锥的研究进展
【摘要】晶状体后圆锥(PLC)是一类以晶状体后囊膜呈球状或圆锥状膨出的先天性晶状体形态异常疾病。大多数为单侧、散发病例,双侧发病者常为常染色体显性遗传或X染色体连锁遗传,迄今发现的基因突变多集中在 EPHA2基因。目前,PLC的发病机制尚不明确,通过PLC的临床表现,研究者提出了相关发病机制假说,即PLC的形成与晶状体后囊膜薄弱、晶状体上皮细胞过度生长、永存玻璃体原始动脉牵引及色素组织牵引有关。与PLC有关的动物模型可大致分为伴有后囊膜膨出的白内障模型和伴有后囊膜破裂的白内障模型。PLC的临床表现多样,主要症状包括白瞳症、眼位不正、畏光以及视力下降等,术前可以通过裂隙灯检查、超声生物显微镜、Pentacam等影像学检查发现晶状体后囊膜的异常形态改变,及术中的特征性表现如"鱼尾"征和"水母"征明确诊断。PLC合并晶状体混浊时以白内障摘除联合后囊膜撕开/切开手术治疗为主,通过植入人工晶状体或戴镜矫正无晶状体眼状态,术后的弱视训练对提高视功能至关重要。
【关键词】先天性白内障;综述;晶状体后圆锥;遗传学特点
基金项目:陕西省重点研发计划(2022SF-067);空军军医大学临床研究项目(2021LC2219)
DOI:10.3760/cma.j.cn115989-20240223-00048
晶状体后圆锥(posterior lenticonus,PLC)是一种以晶状体后囊膜发生进行性、边界清晰的半球状或圆锥状膨出为表现特征的罕见的晶状体发育异常。PLC的发病率为1/100 000,大多数为单眼、散发,双眼发病者多为常染色体显性遗传或X染色体连锁遗传[1,2,3]。PLC可以表现为单纯型,或单基因综合征型遗传病伴发的眼部表型,如唐氏综合征[4]、Lowe综合征[5]、Alport综合征[6]等。早期文献报道双眼PLC的患病率为0%~10%[1,7],但新近Kekunnaya等[8]报道的63例PLC患者中双眼PLC占比达到1/3。PLC的发病机制尚不明确,其临床诊疗尚存在一定的难度。本文就PLC的病因及发病机制、遗传学特点、动物模型、诊断与鉴别诊断、治疗及预后的研究进展进行综述,旨在提高临床医生对PLC的认识及诊治水平。

迄今关于PLC的病因及发病机制的研究十分有限,研究者依据PLC的临床表型提出了以下关于PLC发病机制的假说。
1.1 晶状体后囊膜薄弱假说
1978年,Crouch等[9]在对PLC并发白内障患者的白内障手术中发现,23眼中19眼晶状体后囊膜的凸出部分较其他部位薄弱,术中易发生破裂,推测PLC的发病可能与晶状体后囊膜存在局部固有薄弱区有关。1984年,Khalil等[10]报道1例双侧PLC并发后囊膜下及核性白内障病例的晶状体组织病理学改变,发现发生后圆锥的部位囊膜较薄,后圆锥顶端处的晶状体囊膜厚度是该年龄组正常囊膜厚度的一半,而赤道部的晶状体囊膜厚度是正常组的1.5倍;此外,在晶状体后囊膜下未发现晶状体上皮细胞以及玻璃体动脉或晶状体血管膜残留。推测是由于囊膜薄弱区域内新生的晶状体皮质向后突出,进而拉伸薄弱区的囊膜,导致囊膜向后凸出,相应部位的晶状体纤维排列紊乱,发生局部的晶状体混浊,而赤道部的囊膜较厚且有坚固的悬韧带支撑不发生变形( 图1 )。
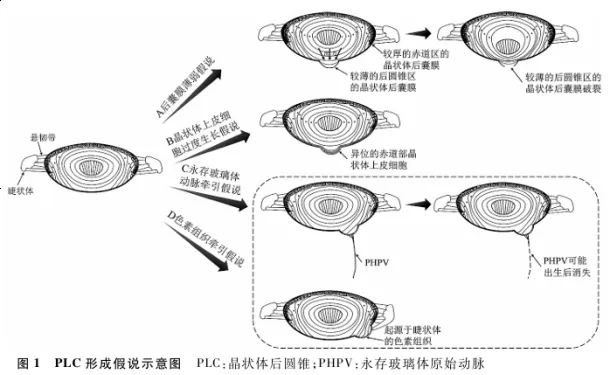
1.2 晶状体上皮细胞过度生长假说
1954年,Franceschetti等[11]报道了1例伴有小角膜、角膜缘发育不良、下方虹膜脉络膜缺损的偏中心的PLC病例,病理检查发现PLC区域的后囊膜下存在许多异位的晶状体赤道部细胞,由此提出晶状体上皮细胞的过度生长并迫使薄弱而有缺陷的晶状体后囊膜向后移位的假说( 图1 )。1955年,Makley等[12]报道了1例视网膜母细胞瘤伴有PLC眼球的病理检查结果,在晶状体后圆锥区域的后囊膜下存在晶状体上皮细胞,上皮细胞在后囊膜凸起的边缘突然停止,在圆锥顶点上方有多层晶状体上皮细胞,而在圆锥边缘附近有单层的细胞,这一发现支持上述晶状体上皮细胞过度生长假说。
1.3 永存玻璃体原始动脉牵引假说
1930年,Butler等[13]在裂隙灯显微镜下观察到一位患者晶状体后囊膜向后膨出,永存玻璃体原始动脉(persistent hyperplastic primary vitreous,PHPV)悬挂在晶状体后方,首次将PLC与PHPV联系起来。1957年,Mann等[14]根据Bulter的报道提出PLC是由于PHPV牵拉晶状体后囊膜形成的。1993年,Kilty等[15]报道了1例单眼PLC伴有PHPV的病例,在晶状体后皮质中央有许多白点和混浊的灰色区域,晶状体后圆锥的尖端连接到一条笔直的灰白色血管上,血管向后延伸至视盘,推测PLC是由玻璃体前动脉和晶状体血管系统异常引起的,这些血管牵引晶状体后囊膜或造成囊膜缺损,随着眼轴长度的增加,对后囊膜的牵引力进一步增强,导致PLC形成。2018年,Khokhar等[16]报道了1例双眼PLC伴后囊膜下白内障的病例,在术中发现细小的PHPV与后囊膜的凸起相连,另一端与视盘相连,证实PLC是由于玻璃体动脉系统的牵引力引起,而对于没有任何PHPV的PLC,推测玻璃体动脉可能在牵拉后囊膜变薄和膨出后已经完全消退( 图1 )。
2002年,Kim等[17]在转录共激活分子的转基因小鼠中发现包括PLC伴白内障在内的多种发育异常,并在2月龄小鼠眼球的病理切片中发现视神经与晶状体的后囊膜之间存在血管连接,因此对PLC的成因也做出与上述相同的推测。
1.4 色素组织牵引假说
目前,关于PLC中色素组织的来源有2种观点。第1种观点认为色素组织来源于初级玻璃体的色素区,2011年,Iwase等[18]报道了1例PLC内及后囊膜周围有色素组织附着的病例,认为色素组织可能起源于初级玻璃体的色素区,参与牵拉并导致PLC。第2种观点认为色素组织来源于睫状体,Agarwal等[19]报道了1例双眼偏中心、伴有色素组织的PLC,同时伴有对应部位的睫状体牵拉,在超声生物显微镜下发现睫状突靠近晶状体,因此推测PLC周围的色素组织可能是晶状体进行性后凸对睫状体造成慢性机械刺激,从睫状体中释放的色素;Zou等[20]报道了1例单眼伴有色素组织的PLC病例,在PLC的组织切片中发现有少量退化的色素上皮细胞,推测色素组织可能来源于虹膜或睫状体色素上皮。
有一种前部型永存胎儿血管(persistent fetal vasculature,PFV)是晶状体后部的纤维血管膜,表现为异常退化的晶状体后血管附着于后囊膜周边部位,纤维血管膜牵拉睫状突,使之向后囊膜中央方向移位,导致睫状突被拉长[21]。结合PFV表型与含色素组织的PLC,推测含色素组织的PLC的形成过程可能是前部型PFV对晶状体后囊膜的牵引形成后圆锥,并且前部PFV的残留痕迹对睫状体的牵拉或刺激引起的色素组织在PLC的后囊膜周围残留( 图1 )。这种假说与PHPV牵引假说有相同的基础,即胎儿期异常退化的眼部血管对后囊膜的牵拉引起后囊膜圆锥状变形( 图1 中方框所示)。
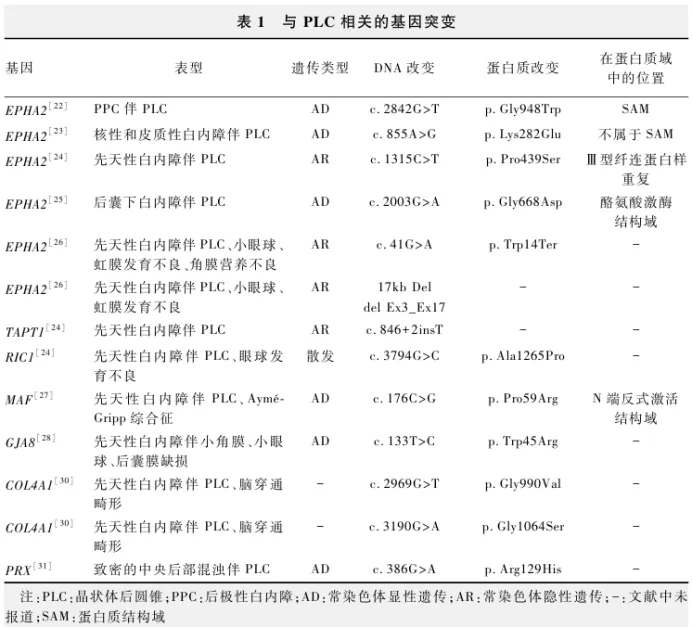
95%的PLC为单眼散发,双眼发病常为常染色体显性遗传或X染色体连锁遗传,双眼PLC伴先天性白内障患者具有明显的遗传异质性。随着遗传学技术的发展,与PLC相关的致病基因也逐渐被发现,对PLC的发病机制探索、基因型-表型的相关性研究、产前筛查及诊断,出生缺陷预防都有很大的帮助。目前,总结与PLC相关的致病基因如下( 表1 )。
2.1 EPHA2基因
Shiels等[22]使用单核苷酸多态性(single nucleotide polymorphism,SNP)标记的全基因组连锁分析对一个后极性白内障(posterior polar cataract,PPC)伴双眼PLC的四代家系进行研究,在 EPHA2基因外显子17中检测到错义变异(c.2842G>T),该基因与疾病共分离,导致翻译水平上甘氨酸替换为色氨酸(p.Gly948Trp),位于SAM结构域,并发现此突变与年龄相关性白内障相关联。Musleh等[23]采用高通量测序(next generation sequencing,NGS)在1例核性白内障伴双侧PLC患者中发现 EPHA2基因外显子4错义变异(c.855A>G),导致翻译水平上赖氨酸替换为谷氨酸(p.Lys282Glu)。Patel等[24]用全外显子组测序(whole-exome sequencing,WES)技术在1例PLC伴先天性白内障患者中发现 EPHA2基因外显子6发生了错义变异(c.1315C>T),导致翻译水平上脯氨酸替换为丝氨酸(p.Pro439Ser),位于Ⅲ型纤连蛋白样重复结构域。Zhai等[25]在1个四代先天性后囊下白内障伴PLC中国家系中发现 EPHA2基因的另一种错义变异(c.2003G>A),位于激酶结构域中,造成受体不稳定,改变了其亚细胞定位,影响 EPHA2及其配体肝配蛋白的激活,且该突变促进细胞迁移并导致白内障形成。Courdier等[26]报道了1例在出生时被诊断为伴有双侧先天性白内障、小眼球、角膜营养不良及虹膜发育不全的单眼PLC病例,通过NGS测序发现了2个杂合突变,包括 EPHA2基因第1个外显子发生了变异(c.41G>A),导致翻译水平上色氨酸进行错义替换(p.Trp14Ter),以及长度为17 kb的1p36.13缺失,涵盖了 EPHA2基因的内含子3至外显子17;此研究表明 EPHA2基因与多种类型的眼部缺陷相关,并且可能参与全眼的发育。
2.2 RIC1基因
Patel等[24]研究发现 RIC1基因和 TAPT1基因与PLC相关联。研究者通过敲低 RIC1,导致 GJA1在缝隙连接处的定位缺陷,影响了缝隙连接的电导率,这可能是 RIC1突变致白内障的一个潜在机制。 RIC1基因编码的蛋白在细胞内胶原蛋白的运输和分泌过程中起关键作用。该基因的突变可能导致胶原蛋白分泌缺陷,进而影响结缔组织的形成。目前没有直接证据表明 RIC1基因突变会导致PLC,作者推测其突变可能通过影响晶状体的结构完整性间接导致类似PLC的表型。
2.3 MAF基因
Javadiyan等[27]报道了1例双侧先天性白内障的患者,其右眼为核性和PPC,左眼为PLC,除此之外还患有感音神经性听力损失、癫痫、短头畸形和扁平脸。该患者的母亲有相同的临床表现,但症状较轻,并诊断该家族患有Aymé-Gripp综合征;通过NGS测序发现,先证者 MAF基因发生了错义变异(c.176C>G),导致翻译水平上脯氨酸错义替换为精氨酸(p.Pro59Arg)。
2.4 GJA8基因
Zhang等[28]报道了1个患有先天性白内障的四代中国家系,先证者被诊断为双侧先天性白内障,伴有小角膜、小眼球和晶状体后囊膜缺损。对先证者的DNA进行WES,发现了 GJA8基因上的1个新的错义变异(c.133T>C),导致翻译水平上色氨酸错义替换为精氨酸(p.Trp45Arg)。PLC通常表现为圆锥形膨出,凸向玻璃体腔,且常伴有局限性薄弱和混浊,最终可能导致晶状体自发破裂[29]。因此我们推测此家系晶状体后囊膜破损的表型可能是PLC的后续表现。
2.5 COL4A1基因
Nau等[30]报道了2例患有双侧PLC的先天性白内障和脑穿通畸形的患者,发现1例患者 COL4A1基因发生了错义变异(c.2969G>T),致翻译水平上甘氨酸替换为缬氨酸(p.Gly990Val);另1例患者 COL4A1基因发生了错义变异(c.3190G>A),致翻译水平上甘氨酸替换为丝氨酸(p.Gly1064Ser)。
2.6 PRX基因
Jones等[31]通过WES在先证者表现为致密的中央后部混浊伴PLC的一家系中发现意义不确定的 PRX基因错义变异(c.386G>A),致翻译水平上精氨酸替换为组氨酸(p.Arg129His)。
目前,几乎没有针对PLC的相关动物模型,但在研究其他眼病的动物模型中,发现有PLC表型的动物模型,以下对相关动物模型作以总结。
3.1 有后囊膜膨出的白内障动物模型
O377是一种患有由 Crbb2基因突变引起的遗传性、进行性白内障小鼠。Ganguly等[32]观察了出生后第1天(postnatal day 1,P1)、P4、P21的野生型、杂合、纯合突变小鼠的眼球,未发现P1野生型和突变型小鼠晶状体之间存在差异;P4杂合子小鼠晶状体中心区域的纤维细胞未完全脱核,纯合子小鼠赤道部的纤维细胞出现变形、分化障碍;P21杂合子小鼠的晶状体出现空泡,晶状体赤道部的细胞停止伸长,纯合子小鼠的晶状体变小、晶状体皮质液化、后囊膜呈圆锥形向后膨出,这种晶状体的表型与PLC的临床表现很相似。该研究结果显示,白内障晶状体上皮细胞中 Capn3的表达增加,钙离子参与了由 Crbb2基因突变和βB2-晶体蛋白功能受损所引发的病理过程。
3.2 有后囊膜破裂动物模型
3.2.1 晶状体发育异常的后囊膜破裂动物模型
Lop10为一种自发性常染色体显性遗传性白内障小鼠,纯合子表现为致密的白内障并伴有小眼畸形,基因突变位于小鼠3号染色体,为 Gja8基因发生了错义变异(c.64C>G),导致翻译水平上甘氨酸替换为精氨酸(p.Gly22Arg)[33]; Lop11是一种自发性常染色体隐性遗传性白内障小鼠,其 Hsf4基因的第9内含子中插入了早期转座元件,进而导致白内障发生[34]; Lop12是一种自发突变、可遗传的白内障小鼠,纯合子表现为不规则的核性白内障,基因突变位于小鼠1号染色体,为 Crygd基因外显子3发生碱基变异(G>A),导致终止密码子的形成,产生含156个氨基酸的截短蛋白质,会改变γ-晶状体蛋白的折叠,导致晶状体混浊,出现晶状体后囊膜向后膨出的过程[35]。
这3种小鼠模型晶状体的组织学改变均显示晶状体分化异常,并在出生后逐渐出现白内障和后囊膜破裂的改变。纯合子 Lop10小鼠在P1时的晶状体有微小的变化,即晶状体后囊膜和前囊膜下偶见囊状细胞分布;P4时晶状体核中央的后部液化,晶状体核开始向后移位,出生后2周的晶状体后囊膜破裂,晶状体内物质被挤出,晶状体后囊膜向后膨出似乎是后囊膜破裂的前奏。 Lop11小鼠在P0.5和P7时,晶状体有更多的纤维细胞脱核出现异常,提示 Hsf4基因在晶状体纤维细胞正常分化中具有重要作用;P12时,小鼠晶状体的皮质和核区出现空泡,并且大多数伴有晶状体的后囊膜破裂,导致晶状体内的物质释放到玻璃体腔内。2月龄的 Lop12/+小鼠表现为致密的核性白内障,3月龄的小鼠眼组织病理学检查发现晶状体皮质通过破裂的后囊膜进入玻璃体,在脱出的晶状体皮质碎片周围出现显著的反应性纤维增生,并伴有局灶性轻度淋巴细胞浸润。
Lop10、 Lop11、 Lop12这3种白内障模型,均为基因变异导致编码的蛋白结构异常,引起了相似的晶状体分化异常和后囊膜破裂的表型,提示晶状体分化异常与后囊膜膨出甚至破裂可能有共同的致病机制。
晶状体破裂性白内障(rupture of lens cataract,RLC)是常染色体隐性遗传的自发突变白内障小鼠模型[36],小鼠P35~P60时晶状体开始自发性混浊,最初的组织学变化是晶状体皮质纤维的不规则肿胀、凝结、变性和碎裂,在P45~P100时晶状体后极部的囊膜出现破裂。对突变基因进行准确定位发现 Dock5基因第15外显子末端有27个碱基的核苷酸缺失,推测该变异可能以某种方式破坏了DOCK5蛋白的稳定性。在成人的晶状体中,DOCK5主要定位在晶状体上皮细胞中,其可能在胚胎晶状体的后缝线形成中起作用,当晶状体内压力超过生长的临界点时,则会发生后极部囊膜的破裂。
3.2.2 伴有其他部位异常的后囊膜破裂白内障动物模型
AND-34 -/-小鼠是Near等[37]通过同源重组获得的白内障模型小鼠。在2~6个成年AND-34 -/-小鼠眼球病理切片中发现晶状体皮质通过破裂的晶状体后囊被挤出,并伴有皮质液化的全白混浊的白内障,除此之外还发现小鼠有异常深的前房和房角粘连(可能是继发性青光眼),葡萄膜外翻,轻到中度的视网膜神经节细胞丢失,以及覆盖在视神经上含色素的视网膜前膜。继发性青光眼似是由于晶状体后囊膜破裂后,晶状体皮质在眼内引起炎症后的改变。追溯眼部的早期变化发现,P3时小鼠的晶状体前部出现明显的空泡化,但没有明显的晶状体囊膜缺陷,也没有晶状体皮质被挤出,提示晶状体囊膜的变化是发生在出生以后。P24时小鼠晶状体出现后囊膜破裂并伴有晶状体皮质被挤出的表现,P33时小鼠赤道部皮质空泡化,并且常伴有皮质被挤出的症状。
Cohen综合征是一种罕见的遗传性疾病,由 VPS13B基因变异引起,是一种以四肢纤细、躯干肥胖、肌张力减退,以及颅面部、口腔、眼部和肢体异常为主要临床特征的出生缺陷。Lhussiez等[38]构建了 Vps13b ΔEx3/ ΔEx3 小鼠模型,并通过眼底镜检查发现小鼠在1~2个月龄(早期)表现为核性白内障,3~5个月龄(晚期)则演变为完全混浊,伴有空泡结构;组织病理学显示,在白内障初期晶状体皮质区出现大空泡,白内障晚期晶状体核通过破裂的后囊膜被挤至玻璃体腔。此外,模型小鼠还伴有视网膜皱褶和水肿,免疫荧光染色显示褶皱和水肿处出现小胶质细胞的激活,这可能是进入玻璃体腔的晶状体物引起的视网膜的炎症反应。雄性模型小鼠还因为少弱畸形精子症而导致不育[39]。
综上所述,后囊膜破裂动物模型可大致分为2类,即主要表现为伴晶状体发育异常的后囊膜破裂动物模型和伴有全身系统异常的后囊膜破裂动物模型。
4.1 临床表现
PLC的临床表现多样,需要结合病史、眼部检查、影像学检查、术中表现做出最终诊断。其常见的2个症状为白瞳症和眼位不正,其次为畏光、视力下降等。大约1/3的早期无白内障改变的PLC患者在裂隙灯显微镜下可出现晶状体后囊膜局限性圆锥状或半球状膨出,红光下呈现"油滴样"反光改变,在出现圆锥的部位表现为近视,而在圆锥的周围通常是远视或正视,导致屈光不正难以矫正,进而导致弱视[40]。也有文献报道在裂隙灯显微镜检查中后囊完整的PLC晚期病例有"环礁"征,即具有完整后囊的透明PLC被混浊的晶状体(砂石)包围[41]。
在大多数PLC病例的诊断年龄在3~7岁[7],首诊主要原因是发现白内障。1978年Crouch等[9]报道了21例PLC伴白内障的患者,其中2例为双眼PLC,其余19例为单眼PLC,首诊年龄为3个月~15岁。研究者仅在4例病例中观察到PLC的早期表现,即"油滴样"反光改变,而大多数患者在出现白内障后被后续诊断为PLC。Cheng等[1]对40例PLC患者的41眼进行研究,其中仅1例为双眼PLC,首次诊断的平均年龄为3岁5个月,在5~7岁年龄段患者主要表现为视力下降,因学校体检中发现视力下降被转诊;值得注意的是在这个研究队列中有8例患者,包括1例双侧发病患者,均在1岁前就诊,反映出儿科医生对PLC早期诊断的认识在不断提高。2000年Hosal等[42]回顾性分析了171例接受单侧先天性白内障摘除术的患者的临床记录,其中19例伴有PLC,PLC诊断的年龄中位数为15个月,8例(42.1%)术后视力达到6/12或更好,4例(22.1%)获得了良好的双眼视功能。Kekunnaya等[8]对63例PLC患者的84只眼进行了研究,其中单眼患病为42例(66%),双侧患病为21例(33%),单眼PLC的平均首诊年龄在6岁6个月,而双眼PLC的平均首诊年龄在3岁,出现这种差异的原因可能与双眼患病对视力、生活及学习的影响大于单眼,患儿的父母更易察觉有关。
综上,对PLC的早期诊断主要依赖于对婴幼儿的眼筛查,以及医生对PLC体征的认识;在出现白瞳、斜视等明显的眼部并发症之前,视力下降或视力差是在眼部健康筛查时容易被较早发现的原因。当出现白瞳症以后,PLC的确诊多依赖于在术中发现了晶状体的后囊膜相应改变。
4.2 影像学检查
已发生白内障或视轴区混浊时,需借助影像学检查进行诊断。B型超声在临床上是重要且经常使用的影像学检查,它可显示PLC的后囊膜膨出,提示晶状体的厚度增加、局部后囊膜缺损、伴或不伴囊膜破裂的隆起,以及玻璃体前部皮质的异常情况。
超声生物显微镜和Scheimpflug图像(Pentacam)也可显示PLC及后囊膜是否有缺损[43]。眼前节光学相干断层扫描可以提供具有较高轴向和横向空间分辨率的眼前节扫描图像,但由于轴向范围有限和对PLC的分辨率相对较低,普通光学相干断层扫描成像可能不足以对变形的晶状体的整个区域进行成像,Chen等[44]使用扫频眼前节光学相干断层扫描对有PLC的晶状体生物参数进行了测量,获得了较完整且清晰的PLC图像,甚至可以检测到细微的PPC,可帮助临床医生进行诊断。
4.3 术中表现
部分临床表现和影像学检查不典型的患者,只有通过仔细的术中观察检查结果方能诊断。有研究描述了PLC患者白内障术中后囊膜裂开后悬在玻璃体腔中的晶状体皮质似鱼尾,并将其称为"鱼尾"征[45]。也有研究报道在白内障术中发现"水母"征,即后囊膜变薄,典型的乳头状突起塌陷到晶状体的囊袋中,并通过灌注液冲洗可重新形成乳头状突起的表现[46]。
临床上与PLC相鉴别的先天性眼部疾病主要有PFV、PPC和后囊膜缺损。
5.1 PFV
PFV是一组因眼部胚胎血管异常退化而导致的疾病,分为前部型、后部型及混合型。通常为单眼发病,散发且不具有遗传性。前部型存在晶状体后部混浊、睫状突拉长或白内障;后部型存在以下一项或多项特征:玻璃体膜、视网膜皱襞或视网膜发育不良、视网膜脱离或视神经发育不全。混合型PFV同时存在前部型和后部型异常,并通过一条贯穿玻璃体腔的纤维条索相连[47]。前部型和混合型PFV常伴有先天性白内障,在摘除混浊晶状体后,可以发现后囊膜有机化、有鬼影血管或后囊膜后凸等异常。少数PLC可与PFV伴发,或者PFV是PLC的部分病因。
5.2 PPC
PPC表现为由畸形晶状体纤维形成中央盘状晶状体混浊,且密集粘附于后囊膜中央。通常为常染色体显性遗传,但也有散发的病例报道。根据报道,有5个基因与PPC直接有关[48],分别为位于染色体1p36的后极性白内障相关基因1( CTTP1)、位于染色体11q22-q22.3的 CTTP2、位于染色体20p12-q12的 CTPP3、位于染色体10q25的 CTPP4、位于染色体14q22-q23的 CTPP5。此外, CRYAB、 CRYGD、 CHMP4B、 MIP、 PITX3、 GJA3、 EPHA2等基因也与PPC有关。PPC不伴有后囊膜的形态异常。
5.3 后囊膜缺损
后囊膜缺损可单眼或双眼发病,常并发于先天性白内障、PLC、PFV、机械性眼外伤等眼部疾病。在术中根据晶状体皮质的抽吸期间或之后发现后囊膜中存在的非手术导致的缺陷得到证实。分为3种类型:Ⅰ型为玻璃体前部皮质下沉的大缺损;Ⅱ型为后囊中的一簇细小缺损;Ⅲ型并发PFV[49]。
有些较大的PLC在发展过程中发生后囊膜破裂,后囊膜形成横椭圆形裂口,晶状体突然发生全白混浊,B型超声提示前部玻璃体混浊,晶状体增厚,甚至后囊膜破裂。
对于晶状体混浊区域直径≥3 mm且位于视轴区的患儿,建议进行手术治疗;而对于混浊区域直径<3 mm或不在视轴区或视力下降不明显的患儿可以进行适当的屈光不正矫正、药物扩瞳和弱视治疗[50]。
6.1 非手术治疗
PLC相关的弱视可能是由晶状体混浊遮挡视轴、屈光参差或"油滴样"改变引起的光学畸变。弱视治疗包括屈光矫正、阿托品扩瞳和遮盖法等。Bradford等[51]对51例单眼视力下降的患者进行弱视治疗并随访,平均随访时间为3年11个月,其中15例单眼先天性白内障组中8例遮盖治疗的效果良好,而在8例单眼PLC组中4例患者的治疗效果却不佳,鉴于PLC组中有2例患者出现进行性白内障,且该组人数较少,研究者不认为PLC患者的视力预后较差。Cheng等[1]对40例PLC患者的41眼进行研究,随访时间为13年5个月,发现在白内障术前经非手术方法治疗的22例患者中有9例(41%)的视力有所改善,这9例患者的年龄从5个月到6岁不等;在整个随访期间,除1例患者因诊断为后囊下混浊在视力上无明显变化以外,19(49%)眼术后视力达到20/40~20/20,7(18%)眼达到20/100~20/50,4(10%)眼达到20/200,4(10%)眼低于20/200。Travi等[52]报道24例平均年龄为4岁的PLC伴或不伴白内障的患者均出现弱视,10例PLC患者中4例(67%)患者非手术治疗弱视1~54个月(中位数为4个月)后视力至少改善了0.3 LogMAR。
6.2 手术治疗
当PLC导致无法矫正的视力下降或合并白内障时,必须进行手术摘除晶状体并植入人工晶状体,且手术治疗后,近期视力预后普遍良好[1,52]。后囊膜完整性为人工晶状体提供了稳定的支撑环境,在白内障手术中处于重要地位[53],对于PLC病例,必须检查后囊的完整性并排除后囊膜是否已经破裂[54]。手术方式大多为超声乳化白内障吸除、后囊膜切开及前部玻璃体切除,并根据患儿年龄和眼部发育情况选择一期或二期人工晶状体植入[55]。超声乳化吸除可降低术中后囊膜破裂的风险,但粘弹剂的使用要适量,以防过度使用增加前房压力导致后囊破裂。如果后囊膜破裂,需将人工晶状体植入睫状沟中。
对于已经存在的后囊破裂,可以通过扩瞳观察到后囊膜的缺损边缘、后囊膜上的白点和"鱼尾"征来诊断PLC。然而,先天性白内障的多种表现导致术前难以对后囊膜缺损做出诊断。Li等[56]发现有后囊膜缺损眼与正常眼的角膜直径存在显著差异。在不明确后囊膜有无缺损的情况下手术,术中发生后囊膜破裂等并发症的风险更高。因此,更全面地总结术前线索以预测是否已经存在后囊膜缺损非常重要。
如果能及时正确地处理,PLC患者的预后通常良好。在后囊膜破裂的病例中,如果处理得当,将人工晶状体植入睫状沟中,通常预后也良好。决定PLC患者视力预后的因素尚不确定。有文献报道诊断年龄与预后视力结果显著相关[7];但Travi等[52]和Jain等[57]均未发现就诊年龄与视力之间存在相关性。Jain等[57]和Wilson等[45]发现视力结果与术前是否斜视之间有相关性。Chen等[58]分析病变的后投影长度和平均角膜曲率是最终视力改善程度的预测因素,角膜不规则程度越低,视力改善越明显。此外,术后屈光矫正和弱视训练对于术后长期预后效果至关重要,需要眼科医师、视光医师和患儿父母互相配合、长期合作,以期为患儿带来更优视力预后。
综上所述,PLC是一类先天性晶状体形态异常疾病,绝大多数为单眼散发病例,双眼病例常为家族遗传,病因及发病机制目前不明。PLC临床表现形式多样、隐匿、易误诊和漏诊,及时的诊断、正确的治疗以及弱视训练对最终获得良好的视功能尤为重要。